crf即CRF,中文意思是病例报告表,指要按照试验方案要求去设计,然后向申办者报告的记录受简举空试者去提供相关信息的纸质或者电子文件。
向你们补充一下另外一个相似的名词eCRF specification即电子病例报告表说明,我们通常说成eCRF Spec。
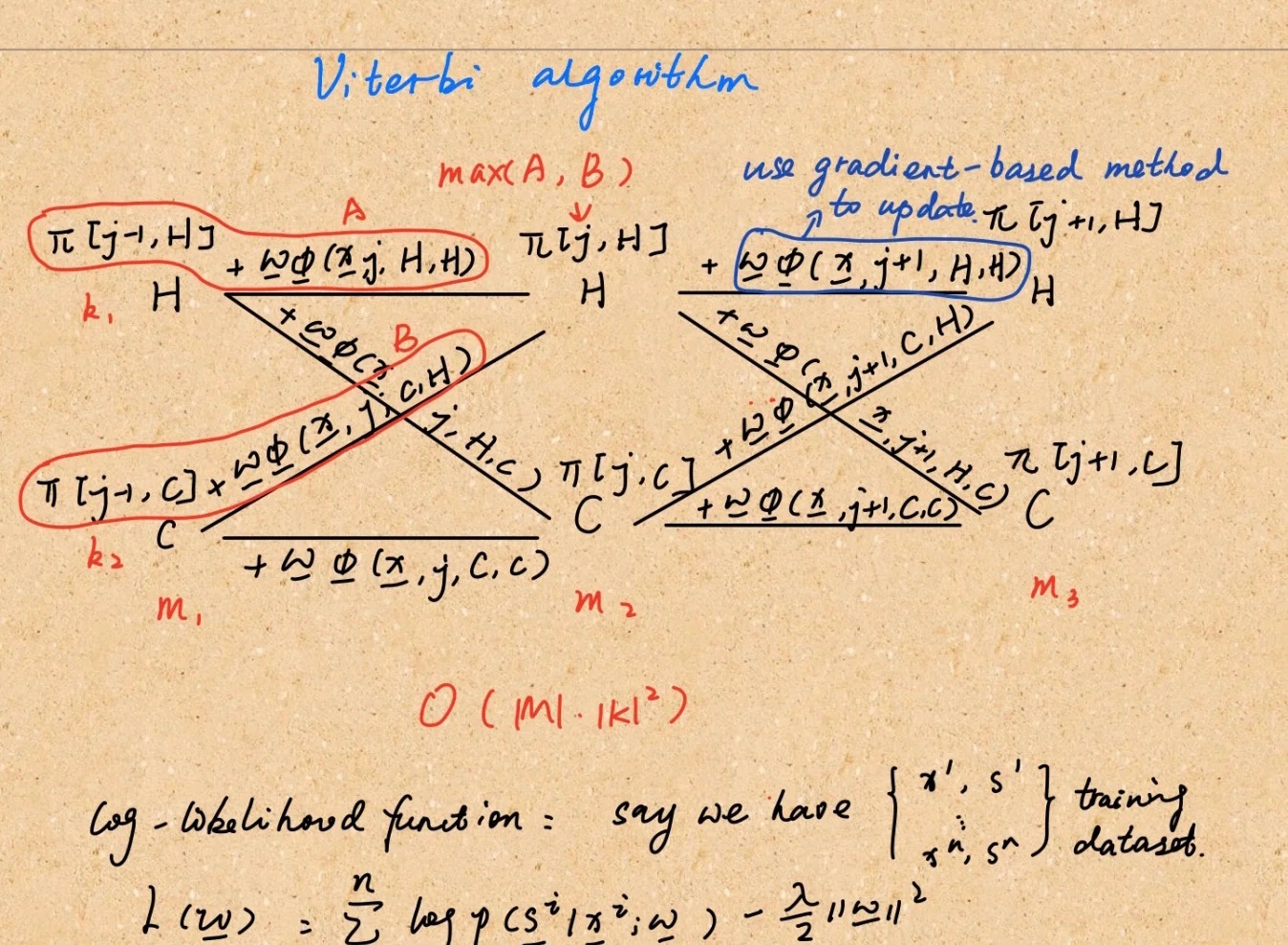
DM在收到最终版临床试验方案后就可以开始撰写eCRF Spec。
一般构成:目录、修订历史、时间节点、specificform(例 enroll、SV、ICF等)
目录:一般与EDC中的顺序一致
修订历史:版本号(例如,版本1.0到2.0)。DM将详细记录对 spec的变更。
所以希望有些朋友不要把这两个都弄混了!
不仅仅是报告表,中期也是监查阶段,是CRA工作的核心!
当然要做的工作只是包括这些但不限于这些,就如以下四点:
(一)SDV:核对病历与CRF的一致性逻辑性,保证
收集数据真实性、完整、准确(CRF是病例报告表)
(二)药品管理:核查药品供应、存储、分发、回收、销毁(这里会设计药品发放表,回收表,库存表及销毁表灯多种文件,要记得好好存档哟)
(三)文件管理:保证临床试验活动产生的所有文件归档到相应文件夹当中(无拦瞎数据即无发生)
(四)安全性管理:确保AE/SAE的及时识别、记录、上报(伦理递交的文件及时取回并扫描保存)临床试验数据都是在监查阶段产生,所以这一答慧阶段的数据要仔细核对,保证数据的准确性,完整性,才能更好的保障受试者的权益。
这些也都是CRF的管理范围!


